 hepacentral.com
hepacentral.com
Liver fibrosis represents one of the most complex and clinically significant pathological processes in hepatology, characterized by the excessive accumulation of extracellular matrix components that fundamentally alter hepatic architecture and function. This intricate biological phenomenon serves as the common pathway through which diverse chronic liver injuries ultimately progress toward cirrhosis, portal hypertension, and hepatocellular carcinoma. Understanding the molecular mechanisms and cellular interactions that drive fibrogenesis has become increasingly critical as the global burden of chronic liver disease continues to escalate, with conditions ranging from viral hepatitis and alcoholic liver disease to non-alcoholic fatty liver disease affecting hundreds of millions of individuals worldwide.
The pathogenesis of liver fibrosis encompasses a remarkably orchestrated cascade of molecular events that transforms the liver’s normal regenerative response into a pathological process of progressive scarring. This transformation involves the coordinated activation of multiple cell types, the dysregulation of numerous signaling pathways, and the profound alteration of the hepatic microenvironment. Rather than representing a simple accumulation of scar tissue, liver fibrosis emerges from the complex interplay between chronic injury, persistent inflammation, aberrant wound healing responses, and the disruption of normal tissue homeostasis.
The Cellular Orchestra of Fibrogenesis
The development of liver fibrosis requires the participation of virtually every cell type within the hepatic parenchyma, each contributing unique functions that collectively drive the fibrogenic process. Hepatic stellate cells occupy the central position in this cellular orchestra, serving as the primary source of extracellular matrix proteins and the key effector cells responsible for the structural changes that define fibrotic tissue. In their quiescent state, these perisinusoidal cells function as the liver’s primary vitamin A storage repository, maintaining hepatic retinoid homeostasis while remaining largely inactive with respect to extracellular matrix production.
The transformation of quiescent hepatic stellate cells into activated myofibroblasts represents perhaps the most critical cellular event in fibrogenesis. This phenotypic transition involves profound changes in gene expression, cellular morphology, and functional capacity. Activated stellate cells lose their characteristic lipid droplets containing vitamin A, develop prominent stress fibers composed of alpha-smooth muscle actin, and dramatically upregulate the production of collagens, particularly types I and III. This activation process occurs in response to a complex array of stimuli, including growth factors, cytokines, oxidative stress, and direct cell-cell interactions.
Hepatocytes, as the liver’s primary parenchymal cells, play multifaceted roles in fibrogenesis that extend far beyond their traditional metabolic functions. Injured hepatocytes serve as both targets and perpetrators of the fibrogenic process, releasing damage-associated molecular patterns that trigger inflammatory responses while simultaneously producing profibrogenic mediators. The death of hepatocytes through apoptosis, necrosis, or pyroptosis creates signals that directly activate stellate cells and recruit inflammatory cells to sites of injury. Additionally, surviving hepatocytes can undergo epithelial-mesenchymal transition under certain circumstances, contributing directly to the pool of matrix-producing cells.
Portal fibroblasts represent another crucial cellular component, particularly in cholestatic forms of liver injury where bile duct damage predominates. These cells, which normally maintain the structural integrity of portal tracts, become activated in response to cholestatic injury and contribute significantly to the periportal fibrosis that characterizes diseases such as primary sclerosing cholangitis and primary biliary cholangitis. The activation of portal fibroblasts occurs through mechanisms that are both similar to and distinct from those governing stellate cell activation, involving specific responses to bile acid toxicity and cholangiocyte-derived signals.
Liver sinusoidal endothelial cells occupy a unique position at the interface between the hepatic vasculature and the space of Disse, making them critical regulators of stellate cell activation and quiescence. In healthy liver tissue, these specialized endothelial cells maintain stellate cell quiescence through the continuous production of nitric oxide and other anti-fibrogenic mediators. However, chronic liver injury leads to endothelial dysfunction characterized by capillarization, loss of fenestrations, and reduced nitric oxide production. This endothelial dysfunction not only permits stellate cell activation but also promotes the development of intrahepatic vascular resistance that contributes to portal hypertension.
Molecular Mediators and Signaling Networks
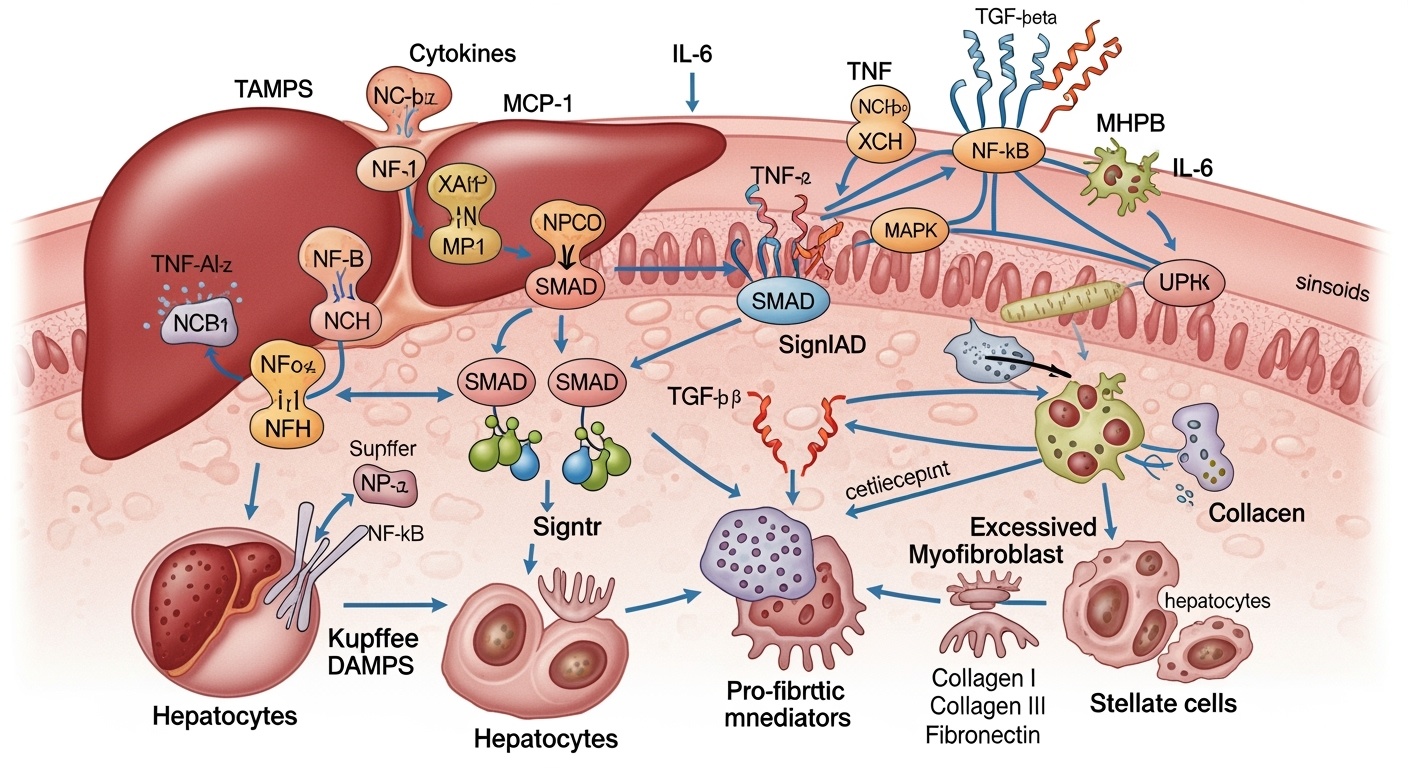
The molecular landscape of liver fibrosis encompasses an extraordinarily complex network of signaling pathways, growth factors, cytokines, and transcriptional regulators that coordinate the cellular responses underlying fibrogenesis. Transforming growth factor-beta stands as the most potent and well-characterized profibrogenic mediator, orchestrating virtually every aspect of the fibrogenic response. This multifunctional cytokine promotes stellate cell activation, enhances extracellular matrix synthesis, inhibits matrix degradation, and suppresses hepatocyte proliferation. The TGF-β signaling pathway operates through both canonical Smad-dependent mechanisms and non-canonical pathways involving MAP kinases, PI3K/Akt, and other signaling cascades.
The regulation of TGF-β activity represents a critical control point in fibrogenesis, involving complex mechanisms of ligand activation, receptor expression, and downstream signal transduction. TGF-β is secreted in a latent form that requires activation through mechanical forces, proteolytic cleavage, or interaction with specific activating proteins such as thrombospondin-1 and integrin αvβ6. Once activated, TGF-β binds to its receptors and initiates signaling cascades that culminate in the transcriptional activation of profibrogenic genes including collagens, tissue inhibitors of metalloproteinases, and alpha-smooth muscle actin.
Platelet-derived growth factor represents another crucial mediator of stellate cell activation, serving as a potent mitogen and chemoattractant for these cells. PDGF signaling promotes stellate cell proliferation, migration, and contractility, contributing to both the expansion of the activated stellate cell population and their ability to contract and distort hepatic architecture. The PDGF pathway intersects with numerous other signaling networks, including those mediated by TGF-β, creating complex regulatory circuits that amplify and sustain the fibrogenic response.
The role of inflammatory mediators in liver fibrosis extends beyond simple tissue damage to encompass direct profibrogenic effects on stellate cells and other liver cell types. Interleukin-1β, tumor necrosis factor-α, and interleukin-6 not only promote hepatocyte injury and inflammatory cell recruitment but also directly stimulate stellate cell activation and matrix production. The complement system, traditionally associated with antimicrobial defense, has emerged as an important contributor to liver fibrosis through its ability to promote inflammatory cell activation and stellate cell stimulation.
Oxidative stress represents both a consequence and a driver of liver fibrosis, creating self-perpetuating cycles that maintain and amplify the fibrogenic response. Reactive oxygen species generated by damaged hepatocytes, activated Kupffer cells, and infiltrating neutrophils directly activate stellate cells while also promoting the production of profibrogenic mediators. The antioxidant systems normally present in the liver become overwhelmed during chronic injury, leading to persistent oxidative stress that perpetuates cellular damage and fibrogenic signaling.
Extracellular Matrix Dynamics and Remodeling
The pathological accumulation of extracellular matrix that defines liver fibrosis results from profound alterations in the balance between matrix synthesis and degradation. Normal liver tissue contains a minimal amount of interstitial collagen, primarily located in portal tracts and around central veins, with the space of Disse containing a specialized matrix composed of low-density collagen type IV, laminin, and proteoglycans. The development of fibrosis involves both quantitative and qualitative changes in matrix composition, with a dramatic shift toward high-density fibrillar collagens that fundamentally alter hepatic architecture and function.
Collagen type I emerges as the predominant matrix protein in fibrotic liver tissue, forming thick bundles that create the characteristic bridging fibrosis observed in advanced disease. The synthesis of collagen type I involves complex post-translational modifications including hydroxylation of proline and lysine residues, formation of stable cross-links, and assembly into mature fibrils. The regulation of collagen synthesis occurs at multiple levels, including transcriptional control by TGF-β and other growth factors, post-transcriptional regulation by microRNAs, and post-translational modifications that affect protein stability and function.
The degradation of extracellular matrix represents an equally important component of matrix homeostasis, mediated primarily by matrix metalloproteinases and their endogenous inhibitors, the tissue inhibitors of metalloproteinases. In healthy liver tissue, MMP activity maintains matrix turnover and prevents pathological accumulation. However, chronic liver injury leads to a dramatic shift in the MMP/TIMP balance, with increased production of TIMPs and decreased MMP activity resulting in net matrix accumulation. This imbalance is particularly pronounced for MMP-1, the primary collagenase responsible for degrading fibrillar collagens.
The cross-linking of collagen fibers represents a critical determinant of matrix stability and the reversibility of fibrosis. Lysyl oxidase and lysyl hydroxylase catalyze the formation of aldol condensation products and pyridinoline cross-links that stabilize collagen fibers and resist proteolytic degradation. The activity of these cross-linking enzymes increases during fibrogenesis, contributing to the progressive stabilization of fibrotic tissue and the decreased likelihood of spontaneous resolution.
Matrix stiffness emerges as both a consequence and a driver of fibrogenesis, creating mechanobiological feedback loops that perpetuate the fibrotic process. As the liver becomes increasingly fibrotic, the mechanical properties of the tissue change dramatically, with increased stiffness providing signals that further activate stellate cells and promote continued matrix synthesis. This mechanotransduction occurs through integrin-mediated signaling pathways and the activation of mechanosensitive transcription factors such as YAP/TAZ.
Inflammatory Networks and Immune Cell Participation
The inflammatory component of liver fibrosis encompasses far more than simple tissue damage, involving sophisticated networks of immune cells, inflammatory mediators, and tissue-resident cells that collectively orchestrate the fibrogenic response. Kupffer cells, the liver’s resident macrophages, occupy central positions in these inflammatory networks, serving as both sensors of tissue damage and effectors of fibrogenic signaling. Upon activation by danger signals, Kupffer cells release numerous profibrogenic mediators including TGF-β, PDGF, and reactive oxygen species while also orchestrating the recruitment of additional inflammatory cells.
The phenotypic plasticity of macrophages adds significant complexity to their roles in liver fibrosis, with different activation states promoting either fibrogenesis or resolution. Classically activated M1 macrophages contribute to tissue injury and early fibrogenic responses through the production of proinflammatory cytokines and reactive oxygen species. In contrast, alternatively activated M2 macrophages can promote both fibrogenesis through TGF-β production and resolution through the secretion of matrix metalloproteinases and phagocytosis of apoptotic cells.
Neutrophils, despite their traditionally recognized role in acute inflammation, contribute significantly to chronic liver fibrosis through multiple mechanisms. These cells release elastase and other proteases that can activate latent TGF-β, generate reactive oxygen species that promote stellate cell activation, and form neutrophil extracellular traps that serve as scaffolds for further inflammatory cell recruitment. The persistence of neutrophil infiltration in chronic liver injury creates a sustained source of profibrogenic signals that maintains stellate cell activation.
T lymphocytes participate in liver fibrosis through both direct effects on stellate cells and indirect effects mediated by cytokine production. Th17 cells, characterized by their production of interleukin-17, promote stellate cell activation and proliferation while also enhancing the recruitment of neutrophils and other inflammatory cells. Regulatory T cells can exert anti-fibrogenic effects through the production of interleukin-10 and direct cell-cell interactions, but their function may be impaired in chronic liver disease.
Natural killer cells contribute to liver fibrosis through their ability to kill activated stellate cells, potentially serving as a natural brake on fibrogenesis. However, chronic liver injury often leads to NK cell dysfunction, reducing their anti-fibrogenic capacity and permitting unchecked stellate cell activation and proliferation.
| Cell Type | Primary Functions in Fibrogenesis | Key Mediators Released | Activation Triggers |
| Hepatic Stellate Cells | Matrix synthesis, contractility, inflammatory signaling | Collagen I/III, α-SMA, TIMP-1, TGF-β | TGF-β, PDGF, oxidative stress, mechanical tension |
| Kupffer Cells | Inflammatory orchestration, stellate cell activation | TGF-β, TNF-α, IL-1β, ROS, PDGF | LPS, damage signals, complement fragments |
| Portal Fibroblasts | Periportal matrix deposition, bile duct support | Collagen I/III, fibronectin, proteoglycans | Bile acids, cholangiocyte signals, TGF-β |
| Liver Sinusoidal Endothelial Cells | Vascular regulation, stellate cell modulation | NO (anti-fibrogenic), VEGF, endothelin-1 | Hypoxia, shear stress, inflammatory cytokines |
Metabolic Reprogramming in Fibrogenesis
The development of liver fibrosis involves profound alterations in cellular metabolism that both drive and sustain the fibrogenic process. Activated hepatic stellate cells undergo dramatic metabolic reprogramming characterized by increased glycolysis, altered amino acid metabolism, and enhanced biosynthetic capacity to support the massive production of extracellular matrix proteins. This metabolic transformation resembles the Warburg effect observed in cancer cells, with stellate cells shifting from oxidative phosphorylation toward glycolysis even in the presence of adequate oxygen.
The increased glycolytic flux in activated stellate cells serves multiple functions beyond simple energy production, providing metabolic intermediates for collagen synthesis and supporting the pentose phosphate pathway that generates reducing equivalents necessary for biosynthetic processes. Glucose-6-phosphate dehydrogenase, the rate-limiting enzyme of the pentose phosphate pathway, becomes markedly upregulated in activated stellate cells, supporting both antioxidant defense and nucleotide synthesis required for cell proliferation.
Lipid metabolism undergoes equally profound changes during stellate cell activation, with the characteristic loss of retinoid-containing lipid droplets accompanied by alterations in fatty acid synthesis and oxidation. The mobilization of stored vitamin A not only serves as a marker of stellate cell activation but also provides retinoic acid that can influence gene expression through nuclear hormone receptors. Simultaneously, activated stellate cells increase fatty acid synthesis to support membrane biosynthesis required for cell proliferation and organelle expansion.
Amino acid metabolism becomes critically important during fibrogenesis due to the enormous demand for proline and hydroxyproline required for collagen synthesis. Stellate cells upregulate enzymes involved in proline biosynthesis, including pyrroline-5-carboxylate synthase and pyrroline-5-carboxylate reductase, while also enhancing the uptake of glutamine and other amino acid precursors. The post-translational hydroxylation of proline residues in collagen requires vitamin C and α-ketoglutarate, linking collagen synthesis to central carbon metabolism.
The metabolic reprogramming of stellate cells creates vulnerabilities that may be exploited therapeutically, with several metabolic pathways representing potential targets for anti-fibrotic intervention. Inhibition of glycolysis, targeting of lipid metabolism, or disruption of amino acid synthesis could theoretically reduce matrix production and stellate cell activation, though such approaches must be carefully balanced against the metabolic needs of healthy liver cells.
Epigenetic Regulation and Gene Expression Control
The stable activation of hepatic stellate cells and the maintenance of the fibrogenic phenotype involve complex epigenetic modifications that alter gene expression patterns without changing the underlying DNA sequence. These epigenetic changes include DNA methylation, histone modifications, chromatin remodeling, and non-coding RNA regulation, creating heritable alterations in gene expression that persist throughout the activated phenotype and may contribute to the persistence of fibrosis even after the removal of injurious stimuli.
DNA methylation patterns undergo significant changes during stellate cell activation, with hypermethylation of promoter regions leading to the silencing of genes associated with quiescence while hypomethylation permits the expression of profibrogenic genes. The peroxisome proliferator-activated receptor gamma gene, which maintains stellate cell quiescence, becomes hypermethylated during activation, contributing to the loss of the quiescent phenotype. Conversely, the promoters of collagen genes become hypomethylated, facilitating their increased expression in activated cells.
Histone modifications represent another crucial layer of epigenetic regulation, with specific patterns of acetylation, methylation, and other post-translational modifications determining the accessibility of genetic loci to transcriptional machinery. Histone deacetylases play particularly important roles in stellate cell activation, with HDAC activity promoting the expression of profibrogenic genes while suppressing anti-fibrogenic pathways. The balance between histone acetyltransferases and deacetylases creates dynamic regulatory systems that can be modulated by therapeutic interventions.
MicroRNAs emerge as critical regulators of fibrogenesis, with specific miRNAs controlling virtually every aspect of the process from stellate cell activation to matrix synthesis and inflammatory cell recruitment. MicroRNA-29 family members function as master regulators of extracellular matrix production, with their downregulation during fibrogenesis permitting increased collagen synthesis. MicroRNA-122, the most abundant miRNA in hepatocytes, influences lipid metabolism and inflammatory signaling, with its dysregulation contributing to the metabolic aspects of liver fibrosis.
Long non-coding RNAs represent an emerging class of regulatory molecules that influence fibrogenesis through diverse mechanisms including chromatin modification, transcriptional regulation, and post-transcriptional control. These molecules can serve as molecular scaffolds for protein complexes, compete with microRNAs for target binding, or directly interact with DNA to influence gene expression. The identification of fibrosis-associated long non-coding RNAs has opened new avenues for understanding the regulatory networks controlling fibrogenesis.
Mechanobiology and Physical Forces
The mechanical properties of liver tissue undergo dramatic changes during fibrogenesis, with increased stiffness, altered cellular architecture, and changed force transmission creating mechanobiological feedback loops that perpetuate and amplify the fibrotic process. Normal liver tissue exhibits relatively low stiffness due to its high cellular content and minimal extracellular matrix, but fibrotic tissue becomes progressively stiffer as matrix accumulates and cross-links form between collagen fibers.
Hepatic stellate cells respond directly to changes in substrate stiffness through mechanotransduction pathways involving integrins, focal adhesions, and the cytoskeleton. When cultured on stiff substrates that mimic fibrotic tissue, stellate cells spontaneously activate and increase matrix production, while soft substrates that resemble normal liver tissue promote quiescence. This mechanosensitive activation occurs through pathways involving the transcriptional regulators YAP and TAZ, which translocate to the nucleus in response to mechanical stress and promote the expression of profibrogenic genes.
The contractile properties of activated stellate cells contribute significantly to the mechanical aspects of liver fibrosis, with these cells exerting forces that distort hepatic architecture and increase tissue stiffness. The expression of alpha-smooth muscle actin and the development of stress fibers enable stellate cells to generate contractile forces that are transmitted through the extracellular matrix to surrounding cells and structures. This contractility not only contributes to the mechanical properties of fibrotic tissue but also influences portal pressure and hepatic blood flow.
Matrix stiffness affects not only stellate cells but also hepatocytes, endothelial cells, and other liver cell types, creating tissue-wide changes in cellular behavior and function. Hepatocytes cultured on stiff substrates exhibit altered metabolism, reduced synthetic function, and increased susceptibility to injury, suggesting that the mechanical changes associated with fibrosis contribute directly to hepatic dysfunction. Similarly, endothelial cells respond to increased stiffness with changes in barrier function, inflammatory gene expression, and angiogenic capacity.
The irreversibility of advanced fibrosis may be partially attributable to these mechanobiological factors, with the physical properties of heavily cross-linked matrix creating environments that favor continued stellate cell activation even in the absence of ongoing injury. Breaking these mechanobiological feedback loops represents an important therapeutic challenge that may require interventions targeting both the biochemical and physical aspects of fibrotic tissue.
| Fibrogenic Mediator | Primary Source | Target Cells | Mechanism of Action | Therapeutic Targeting |
| TGF-β1 | Kupffer cells, activated HSCs, hepatocytes | HSCs, hepatocytes, immune cells | Smad2/3 phosphorylation, gene transcription | Neutralizing antibodies, receptor antagonists |
| PDGF | Kupffer cells, endothelial cells, platelets | HSCs, portal fibroblasts | Receptor tyrosine kinase, proliferation/migration | Tyrosine kinase inhibitors |
| Angiotensin II | Local RAS activation, systemic circulation | HSCs, Kupffer cells | AT1 receptor, oxidative stress, inflammation | ACE inhibitors, ARBs |
| Endothelin-1 | LSECs, hepatocytes | HSCs, LSECs | GPCR signaling, vasoconstriction, fibrosis | Endothelin receptor antagonists |
Vascular Remodeling and Angiogenesis
The development of liver fibrosis involves not only the accumulation of extracellular matrix but also profound alterations in hepatic vasculature that contribute to portal hypertension and hepatic dysfunction. The normal hepatic microcirculation, characterized by low-pressure sinusoids with fenestrated endothelium, undergoes pathological remodeling that includes capillarization of sinusoids, formation of intrahepatic shunts, and development of aberrant angiogenesis. These vascular changes both result from and contribute to the fibrogenic process, creating complex interactions between matrix deposition and vascular dysfunction.
Sinusoidal capillarization represents one of the earliest and most significant vascular changes in liver fibrosis, involving the loss of endothelial fenestrations, the deposition of basement membrane components in the space of Disse, and the acquisition of continuous endothelial characteristics typical of systemic capillaries. This process occurs in response to chronic injury and inflammatory signals, leading to impaired hepatocyte-sinusoid exchange and contributing to hepatic dysfunction even before significant matrix accumulation occurs.
The formation of basement membrane in the space of Disse involves the deposition of collagen type IV, laminin, and fibronectin, creating a diffusion barrier that impairs the exchange of nutrients, toxins, and other molecules between hepatocytes and sinusoidal blood. This barrier formation contributes to hepatocyte dysfunction and may promote further injury by interfering with normal metabolic processes and detoxification pathways.
Angiogenesis, the formation of new blood vessels, occurs extensively in fibrotic liver tissue but paradoxically contributes to rather than ameliorates tissue dysfunction. The new vessels formed during fibrogenesis are often structurally abnormal, poorly organized, and functionally inadequate, creating arteriovenous shunts that bypass hepatocytes and contribute to portal hypertension. Vascular endothelial growth factor, produced by hypoxic hepatocytes and activated stellate cells, drives much of this pathological angiogenesis.
The development of intrahepatic vascular resistance represents a crucial consequence of fibrosis-associated vascular remodeling, contributing directly to portal hypertension and its complications. This increased resistance results from both mechanical compression of vessels by fibrotic tissue and active vasoconstriction mediated by endothelin-1, angiotensin II, and other vasoactive mediators produced by activated stellate cells and other liver cells.
Resolution Mechanisms and Therapeutic Implications
Despite the traditional view of liver fibrosis as an irreversible process, accumulating evidence demonstrates that fibrotic tissue can undergo resolution through the coordinated activation of anti-fibrotic mechanisms, the apoptosis or inactivation of activated stellate cells, and the degradation of accumulated extracellular matrix. Understanding these resolution mechanisms has profound implications for therapeutic intervention, suggesting that even advanced fibrosis may be amenable to treatment strategies that promote natural resolution pathways.
The spontaneous resolution of liver fibrosis occurs most readily when the underlying cause of liver injury is eliminated, allowing the liver’s natural healing mechanisms to predominate over continued fibrogenesis. Successful antiviral therapy for hepatitis B and C provides compelling clinical examples of fibrosis resolution, with significant reductions in fibrosis stage occurring over months to years following viral suppression. Similarly, abstinence from alcohol in patients with alcoholic liver disease can lead to substantial improvements in fibrosis, though the extent of reversibility depends on the degree of established cirrhosis.
Matrix metalloproteinases play central roles in fibrosis resolution by degrading accumulated collagen and other matrix proteins. The reactivation of collagenolytic activity requires not only increased MMP expression but also decreased production of tissue inhibitors of metalloproteinases, shifting the protease-antiprotease balance toward matrix degradation. MMP-13, in particular, demonstrates potent collagenolytic activity against the type I collagen that predominates in fibrotic tissue, making it a key effector of resolution.
The fate of activated stellate cells during fibrosis resolution remains an area of active investigation, with evidence supporting both apoptotic cell death and phenotypic reversion to quiescence. Natural killer cells contribute to stellate cell apoptosis through direct cytotoxic effects, while changes in the cytokine environment can promote the transition from activated back to quiescent phenotypes. The relative importance of these mechanisms may vary depending on the stage of fibrosis and the specific conditions promoting resolution.
Macrophage polarization toward anti-inflammatory M2 phenotypes appears crucial for fibrosis resolution, with these cells producing matrix metalloproteinases, phagocytosing apoptotic stellate cells, and creating anti-inflammatory microenvironments that favor tissue repair over continued fibrogenesis. Therapeutic strategies aimed at promoting M2 macrophage polarization represent promising approaches for enhancing natural resolution mechanisms.
The identification of endogenous resolution mechanisms has led to the development of therapeutic strategies aimed at mimicking or enhancing these natural processes. Approaches under investigation include the direct administration of matrix metalloproteinases, the use of MMP-inducing agents, strategies to promote stellate cell apoptosis, and interventions designed to shift the inflammatory balance toward resolution. While these approaches show promise in experimental models, their translation to clinical practice requires careful consideration of safety and efficacy in human patients.
Therapeutic Targeting and Future Directions
The complex pathogenesis of liver fibrosis presents numerous potential targets for therapeutic intervention, ranging from the prevention of initial injury to the promotion of established fibrosis resolution. Current therapeutic approaches can be broadly categorized into those that target the underlying cause of liver injury, those that interrupt specific pathways involved in fibrogenesis, and those that promote the natural resolution of established fibrosis.
Antioxidant therapy represents one of the most extensively studied approaches to anti-fibrotic intervention, based on the central role of oxidative stress in stellate cell activation and fibrogenic signaling. Vitamin E, N-acetylcysteine, and other antioxidants have shown efficacy in experimental models of liver fibrosis, though clinical trials have yielded mixed results. The challenge lies in achieving adequate tissue levels of antioxidants while avoiding interference with normal cellular processes that require reactive oxygen species for proper function.
Targeting specific growth factor pathways offers another promising therapeutic avenue, with particular attention focused on TGF-β signaling due to its central role in fibrogenesis. Strategies under investigation include neutralizing antibodies against TGF-β, small molecule inhibitors of TGF-β receptors, and antisense oligonucleotides targeting TGF-β mRNA. However, the pleiotropic effects of TGF-β on immune function, wound healing, and other physiological processes raise concerns about the safety of systemic TGF-β inhibition.
The renin-angiotensin system has emerged as an unexpected but important target for anti-fibrotic therapy, with angiotensin-converting enzyme inhibitors and angiotensin receptor blockers showing anti-fibrotic effects in both experimental and clinical studies. These agents appear to work through multiple mechanisms, including the reduction of oxidative stress, the modulation of stellate cell activation, and the improvement of hepatic hemodynamics.
Modulation of matrix metalloproteinase activity represents a direct approach to enhancing fibrosis resolution, though the challenge lies in selectively enhancing collagenolytic activity while avoiding the degradation of normal extracellular matrix required for tissue integrity. Strategies under investigation include the selective delivery of MMPs to fibrotic tissue, the use of MMP-inducing agents, and the targeted inhibition of specific TIMPs that block resolution.
The development of stellate cell-specific therapeutic targets has benefited from advances in understanding stellate cell biology and the identification of surface markers and signaling pathways unique to activated cells. Approaches under investigation include the use of stellate cell-targeted drug delivery systems, the development of selective stellate cell toxins, and the modulation of stellate cell-specific signaling pathways.
| Therapeutic Approach | Mechanism of Action | Development Stage | Key Challenges |
| TGF-β Antagonism | Neutralizing antibodies, receptor inhibitors | Phase I/II trials | Pleiotropic effects, immune suppression |
| MMP Enhancement | Increased collagenolysis, matrix degradation | Preclinical | Tissue selectivity, normal matrix preservation |
| Stellate Cell Targeting | Cell-specific drug delivery, selective toxicity | Preclinical | Target specificity, off-target effects |
| Metabolic Modulation | Altered cellular energetics, reduced matrix synthesis | Early preclinical | Metabolic complexity, systemic effects |
The future of anti-fibrotic therapy likely lies in combination approaches that target multiple aspects of the fibrogenic process simultaneously, potentially achieving greater efficacy while minimizing the risks associated with targeting any single pathway. The development of such combination therapies will require a deeper understanding of the interactions between different fibrogenic pathways and the identification of optimal therapeutic windows for intervention.
Personalized medicine approaches may also play increasingly important roles in anti-fibrotic therapy, with genetic and molecular profiling used to identify patients most likely to benefit from specific interventions. The heterogeneity of fibrotic disease, even within single etiological categories, suggests that individualized treatment strategies may be necessary to achieve optimal outcomes.
The integration of regenerative medicine approaches with anti-fibrotic therapy represents another promising direction, with strategies aimed at replacing damaged hepatocytes while simultaneously reducing fibrotic tissue. Hepatocyte transplantation, stem cell therapy, and tissue engineering approaches are all being investigated as potential components of comprehensive anti-fibrotic treatment strategies.
The pathogenesis of liver fibrosis encompasses remarkable biological complexity, involving the coordinated interaction of multiple cell types, signaling pathways, and regulatory mechanisms that collectively transform the liver’s normal wound healing response into a pathological process of progressive scarring. The molecular mechanisms underlying this transformation continue to yield insights that inform the development of new therapeutic approaches, offering hope for the millions of patients affected by chronic liver disease worldwide. As our understanding of fibrogenesis deepens, the prospect of effective anti-fibrotic therapy becomes increasingly realistic, potentially transforming the prognosis for patients with chronic liver disease and reducing the global burden of liver-related morbidity and mortality.

